Show code cell content
import mmf_setup;mmf_setup.nbinit()
import logging;logging.getLogger('matplotlib').setLevel(logging.CRITICAL)
%matplotlib inline
import numpy as np, matplotlib.pyplot as plt
This cell adds /home/docs/checkouts/readthedocs.org/user_builds/physics-581-the-standard-model/checkouts/latest/src to your path, and contains some definitions for equations and some CSS for styling the notebook. If things look a bit strange, please try the following:
- Choose "Trust Notebook" from the "File" menu.
- Re-execute this cell.
- Reload the notebook.
How to Solve the (Radial) Schrödinger Equation#
Here we show how to accurately solve the radial Schrödinger equation numerically with long-range Coulomb potential:
where \(V(r) \rightarrow -\alpha/r\) for large \(r\).
For now, we restrict our attention to \(d=3\) dimensions, and \(S\)-wave states where \(l=0\) so that \(\nu^2 - 1/4 = 0\). The radial wavefunctions \(u(r)\) thus satisfy a 1D Schrödinger equation:
Since the operator is linear, this is formally an eigenvalue problem in function space:
Strategies#
There are two basic strategies for solving this type of problem: Shooting and diagonalization. The first is to solve an initial value problem (IVP) with a high-precision solver, and then use a root-finder to adjust the eigenvalue \(E\) so that the solution converges to the appropriate boundary condition. The second involves finding a matrix approximation for the Hamiltonian \(\op{H}\) and diagonalizing this matrix. Each method has its pros and cons:
Shooting
(+) Conceptually simple and capable of high accuracy.
(+) Can use high-precision adaptive integrates like
scipy.integrate.solve_ivp()in combination with robust root-finders likescipy.optimize.brentq().(-) Need to shoot for each eigenstate.
(-) Must determine bounds for the energies. This can require extensive book-keeping or clever searching strategies to ensure states are not missed.
(-) Exponentially growing solutions can break numerically.
(-) Can’t shoot from singular points, and can’t shoot to \(\infty\) without complications.
Some of these negatives can be compensated for by changing variables, or shooting from interior points.
Diagonalization
(+) Simple if you know the right basis.
(+) Returns all eigenstates and energies at once.
(+) Can be simple and sometimes accurate if the right basis can be found (esp. if the problem is analytic and spectral methods can be used).
(-) Not highly accurate if the problem is not analytic or a good basis cannot be found.
We start by demonstrating the diagonalization approach with finite differences since it is very easy to implement and works extremely well in some cases. Unfortunately, to do well with Coulomb-like potentials requires finding a good basis, and this is not trivial, so our ultimate strategy will rely on shooting for finding highly accurate solution.
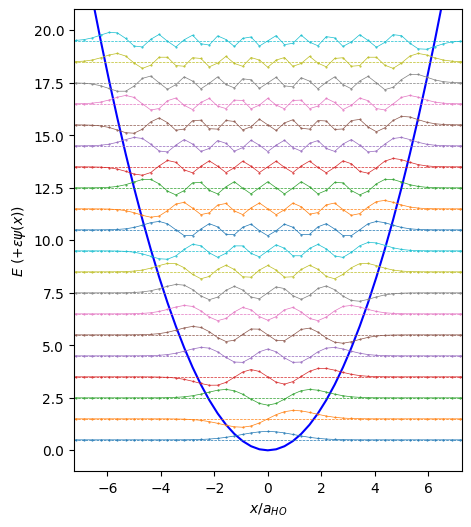
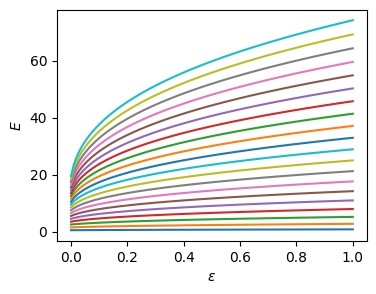
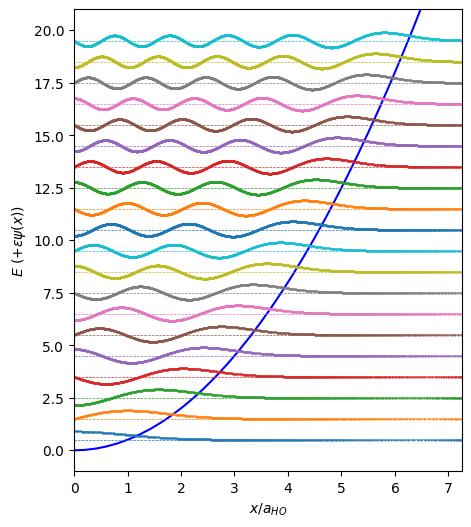
Discretization#
Warning: While this is the simplest approach, it does not work well “out of the box” for the slightly-singular Coulomb potential.
The general approach for solving a linear ODE like this is to discretize the operator by expressing the wavefunction \(u(r)\) in some basis \(\{\ket{f_n}\}\) with basis functions \(f_n(r) = \braket{r|f_n}\):
A convenient basis is that of Dirac delta-functions:
This is the basis for general finite difference methods. The challenge is to find a good representation for the second-derivative operator (Laplacian) in this basis. There are many different approximations that all give the same result in the continuum limit. A common strategy is to use equally spaced abscissa \(r_n = an\) where \(a\) is the lattice spacing. For example, one might use a second-order approximation
This gives the centered difference approximation:
Some care must be taken to implement the correct boundary conditions. These can often be derived with “ghost points”. The form here gives Dirichlet boundary conditions \(u(0) = u(R) = 0\).
Using an appropriate approximation for the derivative one can form the matrix for the Hamiltonian \(\mat{H}\):
where \(\mat{V}\) is a diagonal matrix with \([\vect{V}]_n = V(r_n)\) being the potential evaluated at the lattice sites. We then use a numerical eigensolver to find the eigenstates. Note that this will work quickly and find all of the eigenstates at once, but only converges as a power-law in the spacing \(a\), meaning, that to get the 9 digits of relative accuracy used in [Lepage, 1997], one needs \(a^2 \sim 10^{-9}\), or some \(N \sim 10^5\) lattice points.
Numerov’s Method#
Another closely related method is due to Numerov. This gives an order \(O(a^4)\) approximation, but at the expense of complicating the potential and turning the problem into a generalized eigenvalue problem:
Spectral Methods#
If the potential is smooth and the domain is periodic, then we can often employ a spectral method such as provided by the Fourier transform. This will not be directly relevant for the radial problem (but see the DVR basis), but we include it here as a demonstration. The Laplacian is no-longer tri-diagonal:
Numerical Check#
Here we numerically check these methods, first with a 1D harmonic oscillator, which has a simple analytic solution \(E_n = \hbar \omega (n+\tfrac{1}{2})\) and no singularities, then with the Coulomb potential.
import scipy.linalg
import scipy as sp
# Harmonic oscillator in an optimal box for Fourier methods
# to get 20 states with machine precision.
N = 64
L = 20.0
a = dx = L/N
n = np.arange(N)
x = n*dx - L//2
N_states = 30
hbar = m = w = 1
Vx = m*(w*x)**2/2
En = hbar*w*(np.arange(N) + 0.5) # Exact solution
D2 = (np.diag(np.ones(N-1), k=1)
-2*np.diag(np.ones(N))
+ np.diag(np.ones(N-1), k=-1))
I = (np.diag(np.ones(N-1), k=1)
+ 10*np.diag(np.ones(N))
+ np.diag(np.ones(N-1), k=-1))/12
V = np.diag(Vx)
fig, ax = plt.subplots()
# Finite Difference
H = -hbar**2/2/m * D2/a**2 + V
E, psi = sp.linalg.eig(H)
inds = np.argsort(abs(E))
E, psi = E[inds], psi[:, inds]
ax.semilogy(n[:N_states], abs(E-En)[:N_states], label="Finite Difference")
# Numerov
H = -hbar**2/2/m * D2/a**2 + I@V
E, psi = sp.linalg.eig(H, I)
inds = np.argsort(abs(E))
E, psi = E[inds], psi[:, inds]
ax.semilogy(n[:N_states], abs(E-En)[:N_states], label="Numerov")
# Fourier
# Here is how to compute K by applying the FFT to he identity
k = 2*np.pi * np.fft.fftfreq(N, a)
K = np.fft.ifft(hbar**2*k**2/2/m * np.fft.fft(np.eye(N), axis=1), axis=1)
# Here is the formula from the notes to check.
U = np.exp(2j*np.pi * n[:, None]*n[None, :]/N)/np.sqrt(N)
n_ = (n + N//2) % N - N//2
k = (2*np.pi * n_/ L)
K = U @ np.diag((hbar*k)**2/2/m) @ U.T.conj()
H = K + V
E, psi = sp.linalg.eigh(H)
#inds = np.argsort(abs(E))
#E, psi = E[inds], psi[:, inds]
ax.semilogy(n[:N_states], abs(E-En)[:N_states], label="Fourier")
ax.set(xlabel="$n$", ylabel="Energy Error")
ax.legend();
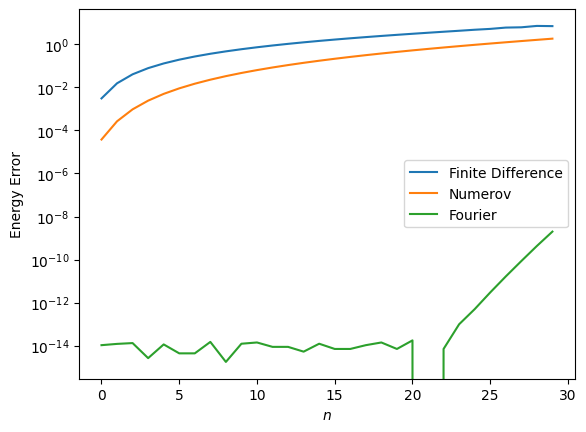
This looks promising, but to really check out code, we should make sure we have the appropriate scaling:
hbar = m = w = 1
L = 20.0
N_states = 20
En = hbar*w*(np.arange(N_states) + 0.5) # Exact solution
Ns = N_states * 2**np.arange(1, 6)
errs = dict(FD=[], Numerov=[])
for N in Ns:
a = dx = L/N
n = np.arange(N)
x = n*dx - L//2
V = np.diag(m*(w*x)**2/2)
D2 = (np.diag(np.ones(N-1), k=1) - 2*np.diag(np.ones(N)) + np.diag(np.ones(N-1), k=-1))
K = -hbar**2/2/m * D2/a**2
I = (np.diag(np.ones(N-1), k=1) + 10*np.diag(np.ones(N)) + np.diag(np.ones(N-1), k=-1))/12
# Finite Differences
E = sp.linalg.eigvalsh(K+V)
errs['FD'].append(abs(E[:N_states]/En - 1))
# Numerov
H = -hbar**2/2/m * D2/a**2 + I@V
E = sp.linalg.eigvals(K+I@V, I)
inds = np.argsort(abs(E))
E = E[inds]
errs['Numerov'].append(abs(E[:N_states]/En - 1))
fig, ax = plt.subplots()
as_ = L/Ns
ax.loglog(as_, errs['FD'], ':o')
ax.loglog(as_, errs['Numerov'], '--+')
ax.plot(as_, as_**2, ':k', lw=4, alpha=0.5, label="$a^2$ (Finite Difference)")
ax.plot(as_, as_**4, '--k', lw=4, alpha=0.5, label="$a^4$ (Numerov)")
ax.set(xlabel="$a=L/N$", ylabel="Relative Energy Error")
ax.legend();
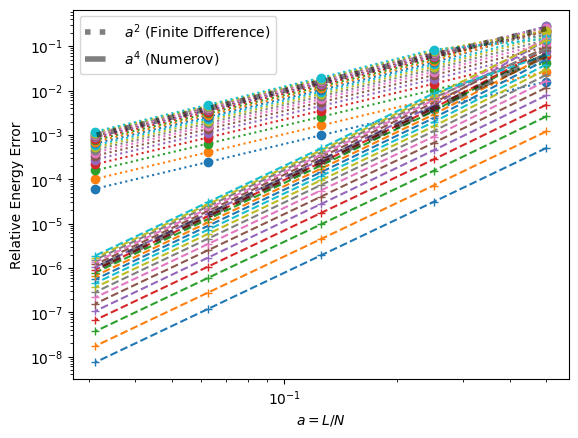
This test confirms that we have correctly implemented the two methods.
Hydrogen Atom#
For testing we use the exact energies for the non-relativistic hydrogen atom:
The eigenstates can also be expressed analytically:
where \(m\) is the reduced mass \(m = m_em_p/(m_e+m_p)\), \(a\) is the reduced Bohr radius, \(L_{n-l-1}^{2l+1}(\rho)\) is a generalized Laguerre polynomial of degree \(n-l-1\), and \(Y_{l}^{m}(\theta, \phi)\) is a spherical harmonic function of degree \(l\) and order \(m\).
These exact solutions might be used to deal with the singular properties of the Coulomb potential, but we postpone discussing this for now in favour of more general techniques.
import scipy.linalg
import scipy as sp
# Hydrogen atom
N = 64*8
R = 20.0
a = R/N
n = np.arange(N)
r = (n + 1)*a
N_states = 30
hbar = m = alpha = 1
Vr = -alpha/r
l = 0
En = -m*alpha**2/hbar**2/2/(1+l+n)**2
D2 = (np.diag(np.ones(N-1), k=1)
-2*np.diag(np.ones(N))
+ np.diag(np.ones(N-1), k=-1))
I = (np.diag(np.ones(N-1), k=1)
+ 10*np.diag(np.ones(N))
+ np.diag(np.ones(N-1), k=-1))/12
V = np.diag(Vr)
fig, axs = plt.subplots(2, 1)
ax = axs[0]
# Finite Difference
H = -hbar**2/2/m * D2/a**2 + V
E, psi = sp.linalg.eig(H)
inds = np.argsort(E.real)
E, psi = E[inds], psi[:, inds]
ax.semilogy(n[:N_states], abs(E/En-1)[:N_states], label="Finite Difference")
print(En[:4])
print(E[:4].real)
axs[1].plot(r, abs(psi[:,0])**2)
axs[1].plot(r, abs(psi[:,1])**2)
# Numerov
H = -hbar**2/2/m * D2/a**2 + I @ V
E, psi = sp.linalg.eig(H, I)
inds = np.argsort(E.real)
E, psi = E[inds], psi[:, inds]
axs[1].plot(r, abs(psi[:,0])**2)
axs[1].plot(r, abs(psi[:,1])**2)
ax.semilogy(n[:N_states], abs(E/En-1)[:N_states], label="Numerov");
print(E[:4].real)
[-0.5 -0.125 -0.05555556 -0.03125 ]
[-0.49980941 -0.12497557 -0.04998816 0.01634702]
[-0.4995119 -0.1249264 -0.04996446 0.01638553]
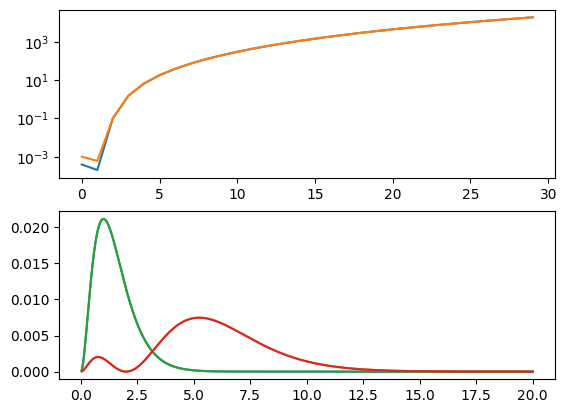
We now see that, although we qualitatively correct solutions, we only have 3 bound states (because our box gets too small), and about 4 places of accuracy, even with a lot of points.
For high accuracy, we probably need to shoot, or be clever.